To align DNA sequences with GATA, first launch GATAliner to create and save the alignment objects. Then launch GATAPlotter to display the alignment. GATAPlotter will initially prompt you for two files, a GATAlignment file generated by GATAliner and an optional gff file describing gene annotation. Use only small region specific gff files, not entire genome gff files.
Additional annotation can be added. See GFF Annotation for detailed documentation.
Here is a key to understanding some of the GATAPlotter displays and features:
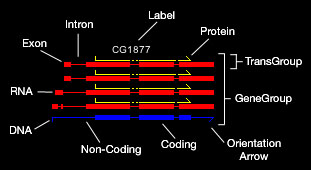
Gene Annotation: The principle component of gene annotation is the GeneGroup. Each GeneGroup is drawn independent of other GeneGroups and allowed
to float within the window to avoid overlap. A typical GeneGroup contains one DNA sequence from
which one or more TransGroups are derived. Each TransGroup contains an RNA transcript and possibly a Protein. Each of these features
are described using standard GFF formatting. It is worth describing how the Coding and Non-Coding DNA sub features
are derived. These are only created in the presence of TransGroups and represent the most conservative estimation of what is protein coding
sequence and what is not protein coding sequence. If any TransGroup predicts a larger coding region than the others, this is adopted for the entire GeneGroup.
Clicking on a GeneGroup will display the complete gene annotation
in the Console window. Arrows indicate orientation. The color, thickness, spacing, and visibility of each feature can be changed using the pull down Annotation menus. These
setting can be saved if so desired, under the File menu, or reset to default values, under the Window menu; so do experiment.
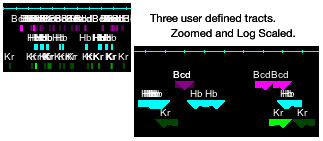
Track Annotation: Unrecognized GFF features are assigned to individual tracks. This is a good place to put non-GeneGroup/ TransGroup annotation like
promoters, locus control regions, enhancers, transcription factor binding sites, etc. Scores associated with these tracked features can be used to scale
their visibility. The larger the score the more opaque. Select which scaling to use in the Annotation menu; linear, log, lnx, or none. Clicking on a
feature prints its associated information in the console window.
DNA Alignments: GATAPlotter uses the alignment objects generated by GATAligner to create two boxes connected by a line. These
boxes are painted onto rectangles representative of the original input sequences. The line connecting the two boxes is colored black for a +/+
oriented alignment or red for a +/- alignment. The boxes and line are also shaded according to their alignment score. Below are two
alignments of Drosophila orthologs showing an inversion and a duplication. The score sliders in the tool panel can be used to dynamically change
which alignment boxes are displayed. To retrieve the alignment information for a particular box, click once on either box or their connecting line.
Information associated with all the visible alignments under the click will be displayed in the console window. Double click the same box or line to
retrieve the actual BLAST local alignment from which the alignment object was derived. Asterisks are used to highlight the alignment object sequence.
Larger sequences can be obtained by dragging the mouse over multiple alignment boxes or selecting the Fetch Conserved Sequences option under the Alignment
menu. Only the visible alignments will be marked as conserved in the fetched sequence.
